REMEDIA challenge
improve health and reduce the overall burden of disease by addressing the impact of environment on health.

REMEDIA stands as a European Human Exposome research project. Its unique methodology relies on using atmospheric simulation chambers, integrating multiple datasets and applying advanced machine learning approaches.
The overall objective of the REMEDIA project is to determine how and to what extent the exposome affects the severity and morbidity of Chronic Obstructive Pulmonary Disease (COPD) and Cystic Fibrosis (CF) throughout the disease, thus providing key elements to design more tailored prevention and care programs.
What is the overall concept underpinning the project?
The rationale of the project is that COPD and CF are two chronic non-communicable diseases that are characterized by a high phenotypic variability of unknown origin, which leads to difficulties in implementing appropriate therapeutic strategies.
The added value of considering both COPD and CF diseases is that, although both are characterized by a similar progressive loss of lung function with small bronchi alterations, the roots of these two diseases are opposite: COPD is currently considered to be mainly related to the external exposome (smoking, ambient particulate matter, household air pollution, occupational particulate matter, ozone and second-hand smoke). While, CF is the consequence of a genetic defect in the CFTR gene, which gives an essential role to factors outside of the exposome. However, modifying genes are now considered to play part in CF disease, therefore also implying a role for internal components of the exposome.
What is an exposome?
Despite the general acknowledgement by the scientific community that ‘Genetics load the gun but environment pulls the trigger’ there is persistent uncertainty as to the global burden of disease attributable to environmental factors.
Throughout our lives, we are exposed to a variety of factors, both internal and external, which are a function of our lifestyle, behavior and environment. All these exposures form a concept called “the exposome”.
The exposome :
The concept of exposome reflects the cumulative measure of environmental influences and associated biological responses from the perinatal period onwards, including exposures from the environment, diet, behavior and endogenous processes. This concept was brought by Christopher Paul Wild in the early 2000s to ‘complement the genome with an exposome’.
Three categories of non-genetic exposures may be considered: internal, specific external and general external.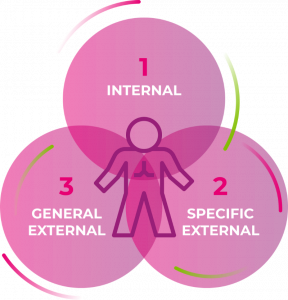
- First, the exposome comprises processes internal to the body such as metabolism, endogenous circulating hormones, body morphology, physical activity, gut microflora, inflammation, lipid peroxidation, oxidative stress and ageing.
- Secondly, there is the extensive range of specific external exposures, which include radiation, infectious agents, chemical contaminants and environmental pollutants, diet, lifestyle factors (e.g. tobacco, alcohol), occupation and medical interventions.
- Thirdly, the exposome includes the wider social, economic and psychological influences on the individual, for example: social capital, education, financial status, psychological and mental stress, urban–rural environment and climate. Here are encompassed the social determinants of health and the ‘causes of the causes’.
Its importance to health and disease is undeniable and the exposome of each individual could completely interact with the way chronic diseases develop and evolve. Air pollution represents a major component of exposome to consider when studying lung diseases
Air pollution:
Environmental exposures are an important component of the external exposome, particularly when considering lung diseases. An adult breathes about 12 m3 of air per day, so the corresponding air quality is of great importance. Air pollution is considered to be the greatest environmental health risk; 9 out of 10 people breathe air containing high levels of pollutants, and about 7 million people die each year from exposure to polluted air. Air pollution is a complex mixture of thousands of different chemical species. One of the main challenges in studying the health effects of air pollution is the extreme complexity of the atmospheric mixture: at any point in space, thousands of pollutants of enormous chemical diversity coexist at concentrations ranging from ppm to ppt, producing immediate chemical reactivity that is impossible to estimate by conventional methods of organic chemistry.
Sources: Who and State of Global Air
Why a focus on lung diseases?
An estimated 12.6 million deaths each year are attributable to unsanitary environments. Of these deaths, 7 million are caused by air pollution leading to chronic respiratory diseases, which are considered one of the leading causes of environment-related deaths.
Among these chronic respiratory diseases, COPD and CF are two highly debilitating diseases that are of particular interest to consider in parallel in a human exposome study. Indeed, the two diseases share common characteristics, including modification of lung function trajectories, yet their roots seem to be opposite: COPD appears to be intricately related to the exposome and CF not.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE (COPD)
COPD is a major non-communicable disease, currently the third leading cause of death in the world, accounting for 5.3% of all deaths, with no treatment available to date. In 2015, 3.2 million people died of COPD worldwide, which represents an increase of 11.6 percent compared with 1990. The annual cost for COPD alone is almost €50 billion, which represents the largest economic burden on health services in terms of respiratory diseases and loss of production in the EU. COPD is characterized by persistent and usually progressive airflow limitation resulting from a combination of a diffuse disease of small airways and the destruction of the pulmonary parenchyma (emphysema). However, COPD patients present multiple variations of their phenotype that may imply different origins and underlying pathophysiological mechanisms, and which increases the complexity of medical treatment.
The main risk factor for developing COPD is smoking; about 40-50% of lifetime smokers will develop COPD. However, at equivalent tobacco consumption, not all smokers develop clinically significant COPD, suggesting that other risk factors should be taken into account, such as occupational exposures, air pollution (indoor/outdoor), as well as genetic factors that may modify the individual risk of developing COPD.
Overall, COPD can no longer be considered as a single entity caused by susceptibility to cigarette smoke, but rather as a syndrome, encompassing several phenotypes. The genetic and environmental determinants identified for a COPD phenotype may not be common to all COPD phenotypes.
Source: Goldcopd
CYSTIC FIBROSIS (CF)
CF is the most common autosomal recessive inherited genetic disorder in the Caucasian population (1 in 2500 births). Although probably under-reported in developing countries, CF affects at least 70,000 persons worldwide, including about 36,000 individuals in the EU. CF places a very heavy burden on people in terms of morbidity, health care utilization, and mortality. In the United Kingdom (U.K.), the average annual cost of CF treatment was between 49,000 and 76,000 euros (in 2012) per patient.
Although advances in basic science and clinical therapeutics have decreased mortality, CF remains an incurable disease and the average life expectancy of a person with CF remains significantly reduced below 50. CF causes lung, pancreatic, digestive and hepatic disorders that express at different levels of severity, therefore leading to a great phenotypic variability of the disease. Obstructive pulmonary disease in CF patients is currently their leading cause of morbidity and accounts for about 80% of mortality. CF is associated with a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, leading to dysfunction of the CFTR protein. To date, more than 2,000 mutations have been identified in the CFTR gene. Importantly, the genotypic variability associated with CF does not fully explain the phenotypic variability. Indeed, although CF is a monogenic disease, there is a wide clinical diversity in patients; the evidence suggests that some manifestations of the disease are related to the severity of the underlying CFTR mutations as well as environmental and genetic modifiers, which currently remain of an overall unknown nature.
Lung function trajectories
Lung function development is critical to lung health at adulthood.
The development of lung function begins in utero and continues throughout childhood, with maximum lung function being reached at the age of 20-25 years. This lung function plateau has long been considered identical to all healthy individuals (i.e. without respiratory symptoms). However, the height of this plateau is now considered highly dependent on events occurring earlier in life; lung function may already be compromised at a very early age (e.g. maternal smoking during pregnancy which results in low birth weight, prematurity, and is associated with poor lung growth and/or broncho-pulmonary dysplasia in the most preterm infants, etc.), and/or by events during childhood and/or adolescence. In addition to the complexity of the global picture, some infants can catch-up lung function while others do not, and there is currently no clue to explain such lung function evolution.
A relatively new concept in COPD pathophysiology is that even if the disease becomes clinically apparent around the age of 40 to 50, its origins are very early in life, namely before birth and in early childhood.
An extended under-standing
The REMEDIA project focuses on the impact of the exposome on the course of two lung diseases: Chronic Obstructive Pulmonary Disease (COPD) and Cystic Fibrosis (CF).
The overall research which will cover the entire life-stages data, will help to detect the most influent exposome components on the development and outcomes of lung diseases through life. To achieve this goal, the REMEDIA project will provide a toolbox allowing the development and tailoring of prevention recommendations and treatment guidelines.
CONTENTS OF THE TOOLBOX:


Recent
news

Publication on Environmental Health Perspectives
Source-specific air pollution and risk of chronic obstructive pulmonary disease: A pooled cohort study Authors: Manuella Lech Cantuaria, Aslak Harbo Poulsen, Ole Raaschou-Nielsen, Étienne Audureau, Ralph Epaud, Sophie Lanone, Jørgen Brandt, Lisa Marie Frohn, Matthias Ketzel, Anja Olsen, Lau Caspar Thygesen, Mette Sørensen Abstract Background: The evidence linking long-term exposure to air pollution and development [...]
Thesis/Dissertation (In French)
Impact des expositions précoces à la pollution atmosphérique sur la maturation pulmonaire et la susceptibilité à développer une BPCO : une étude expérimentale Author: Clément Buissot Abstract La Bronchopneumopathie Chronique Obstructive (BPCO) est une pathologie de l’adulte caractérisée par un déclin progressif de la fonction respiratoire. Issue d’interactions gène-environnement tout au long de la vie, [...]
Publication on Science of The Total Environment
Wildfire and African dust aerosol oxidative potential, exposure and dose in the human respiratory tract Authors: Maria Mylonaki, Maria Gini, Maria Georgopoulou, Marika Pilou, Eleftheria Chalvatzaki, Stavros Solomos, Evangelia Diapouli, Elina Giannakaki, Mihalis Lazaridis, Spyros N. Pandis, Athanasios Nenes, Konstantinos Eleftheriadis, Alexandros Papayannis Abstract Exposure to wildfire smoke and dust can severely affect air quality [...]